Molecular filters#
Molecular filters are predefined sets of conditions, used to remove unwanted molecules, e.g. too large ones. scikit-fingerprints implements many molecular filters, designed for different applications.
They are generally divided into two types, depending on what they use for defining filter conditions:
Physicochemical properties - define allowed numerical ranges of properties like molecular weight, formal charge, or number of rings. A molecule fulfills the filter condition and is kept if all its properties are contained in those ranges.
Molecular substructures - define unwanted substructures, functional groups, toxicity-inducing patterns etc. A molecule fulfills the filter condition and is kept if it does not contain any of the substructures.
Physicochemical properties filters can therefore be thought of as “inclusion” filters, and substructure filters as “exclusion” filters.
Let’s see some examples of both.
Physicochemical filters#
Let’s see a few examples of physicochemical filters:
Molecular weight (docs):
simply checks if a total molecular mass is inside a given range
by default, the range is \([0, 1000]\), corresponding roughly to most small molecules
Lipinski Rule of 5 (docs):
one of the most famous molecular filters, checking for oral bioavailability
tries to select molecules that are small and lipophilic
by default, allows breaking one rule
molecular weight \(\leq 500\)
number of hydrogen bond acceptors (HBA) \(\leq 10\)
number of hydrogen bond donors (HBD) \(\leq 5\)
logP \(\leq 5\)
Beyond Rule of 5 (docs):
designed to cover novel orally bioavailable drugs
less strict than Lipinski filter
molecular weight \(\leq 1000\)
logP in range \([−2, 10]\)
HBA \(\leq 15\)
HBD \(\leq 6\)
topological polar surface area (TPSA) \(\leq 250\)
number of rotatable bonds \(\leq 20\)
Rule of 3 (docs):
optimised to search for fragment-based lead-like compounds with desired properties
there is also an extended rule, including TPSA and rotatable bonds conditions
molecular weight \(\leq 300\)
HBA \(\leq 3\)
HBD \(\leq 3\)
logP \(\leq 3\)
extended rules: TPSA \(\leq 60\), number of rotatable bonds \(\leq 3\)
All filters include the allow_one_violation argument. If True, molecules still pass the filter even if one of their physicochemical properties falls outside the defined range. Vast majority of filters have default value False for this parameter, except for Lipinski filter.
[1]:
import pandas as pd
from rdkit import Chem
from skfp.filters import (
BeyondRo5Filter,
LipinskiFilter,
MolecularWeightFilter,
RuleOfThreeFilter,
)
<frozen importlib._bootstrap>:241: RuntimeWarning: to-Python converter for boost::shared_ptr<RDKit::FilterHierarchyMatcher> already registered; second conversion method ignored.
For starters, let us prepare some example data.
[2]:
smiles = [
"CC(=O)OC1=CC=CC=C1C(=O)O", # Aspirin
"CN1C=NC2=C1C(=O)N(C(=O)N2C)C", # Caffeine
"CC(C)CC1=CC=C(C=C1)C(C)C(=O)", # Ibuprofen
"C[C@H](CCC(=O)O)CC1=CC=CC=C1", # Cholesterol
]
[3]:
mw_filter = MolecularWeightFilter()
lipinski_filter = LipinskiFilter()
beyond_ro5_filter = BeyondRo5Filter()
ro3_filter = RuleOfThreeFilter()
[4]:
mw_filtered_smiles = mw_filter.transform(smiles)
mw_filtered_smiles
[4]:
['CC(=O)OC1=CC=CC=C1C(=O)O',
'CN1C=NC2=C1C(=O)N(C(=O)N2C)C',
'CC(C)CC1=CC=C(C=C1)C(C)C(=O)',
'C[C@H](CCC(=O)O)CC1=CC=CC=C1']
[5]:
lipinski_filtered_smiles = lipinski_filter.transform(smiles)
lipinski_filtered_smiles
[5]:
['CC(=O)OC1=CC=CC=C1C(=O)O',
'CN1C=NC2=C1C(=O)N(C(=O)N2C)C',
'CC(C)CC1=CC=C(C=C1)C(C)C(=O)',
'C[C@H](CCC(=O)O)CC1=CC=CC=C1']
[6]:
beyond_ro5_filtered_smiles = beyond_ro5_filter.transform(smiles)
beyond_ro5_filtered_smiles
[6]:
['CC(=O)OC1=CC=CC=C1C(=O)O',
'CN1C=NC2=C1C(=O)N(C(=O)N2C)C',
'CC(C)CC1=CC=C(C=C1)C(C)C(=O)',
'C[C@H](CCC(=O)O)CC1=CC=CC=C1']
[7]:
ro3_filtered_smiles = ro3_filter.transform(smiles)
ro3_filtered_smiles
[7]:
['CC(=O)OC1=CC=CC=C1C(=O)O', 'C[C@H](CCC(=O)O)CC1=CC=CC=C1']
By default, filters return a subset of molecules. Alternatively, by setting return_indicators=True, they can return a vector of booleans, with True value corresponding to molecules fulfilling the filter rules. This allows more sophisticated analyzes of results, e.g. by saving the results in a dataframe.
[8]:
mw_mask = MolecularWeightFilter(return_indicators=True)
lipinski_mask = LipinskiFilter(return_indicators=True)
beyond_ro5_mask = BeyondRo5Filter(return_indicators=True)
ro3_mask = RuleOfThreeFilter(return_indicators=True)
/home/jakub/PycharmProjects/scikit-fingerprints/skfp/bases/base_filter.py:109: UserWarning: return_indicators is deprecated and will be removed in 2.0, use return_type instead
warnings.warn(
[9]:
data = [
{"name": "Aspirin", "smiles": "CC(=O)OC1=CC=CC=C1C(=O)O"},
{"name": "Caffeine", "smiles": "CN1C=NC2=C1C(=O)N(C(=O)N2C)C"},
{"name": "Ibuprofen", "smiles": "CC(C)CC1=CC=C(C=C1)C(C)C(=O)O"},
{"name": "Cholesterol", "smiles": "C[C@H](CCC(=O)O)CC1=CC=CC=C1"},
{"name": "Glucose", "smiles": "C(C1C(C(C(C(O1)O)O)O)O)O"},
]
df = pd.DataFrame(data)
[10]:
df["mw_filter_pass"] = mw_mask.transform(df["smiles"])
df["lipinski_filter_pass"] = lipinski_mask.transform(df["smiles"])
df["beyond_rule_of_5_pass"] = beyond_ro5_mask.transform(df["smiles"])
df["rule_of_3_pass"] = ro3_mask.transform(df["smiles"])
[11]:
df
[11]:
| name | smiles | mw_filter_pass | lipinski_filter_pass | beyond_rule_of_5_pass | rule_of_3_pass | |
|---|---|---|---|---|---|---|
| 0 | Aspirin | CC(=O)OC1=CC=CC=C1C(=O)O | True | True | True | True |
| 1 | Caffeine | CN1C=NC2=C1C(=O)N(C(=O)N2C)C | True | True | True | False |
| 2 | Ibuprofen | CC(C)CC1=CC=C(C=C1)C(C)C(=O)O | True | True | True | False |
| 3 | Cholesterol | C[C@H](CCC(=O)O)CC1=CC=CC=C1 | True | True | True | True |
| 4 | Glucose | C(C1C(C(C(C(O1)O)O)O)O)O | True | True | False | False |
Substructural filters#
Substructural filters use sets of SMARTS patterns to define unwanted substructures. An example is Brenk filter (docs), designed to filter out molecules containing substructures with undesirable pharmacokinetics or toxicity, e.g. sulfates, phosphates, nitro groups. Other filters from this group often work based on similar principles, but differing in how aggressively they filter the molecules.
[12]:
from skfp.filters import BrenkFilter
brenk_filter = BrenkFilter()
Meanings of filter conditions are available through .get_feature_names_out() method. Generally, they are interpretable names given by creators, rather than raw SMARTS patterns.
[13]:
brenk_filter.get_feature_names_out()
[13]:
array(['>_2_ester_groups', '2-halo_pyridine', 'acid_halide',
'acyclic_C=C-O', 'acyl_cyanide', 'acyl_hydrazine', 'aldehyde',
'Aliphatic_long_chain', 'alkyl_halide', 'amidotetrazole',
'aniline', 'azepane', 'Azido_group', 'Azo_group', 'azocane',
'benzidine', 'beta-keto/anhydride', 'biotin_analogue',
'Carbo_cation/anion', 'catechol', 'charged_oxygen_or_sulfur_atoms',
'chinone_1', 'chinone_2', 'conjugated_nitrile_group',
'crown_ether', 'cumarine', 'cyanamide',
'cyanate_/aminonitrile_/thiocyanate', 'cyanohydrins',
'cycloheptane_1', 'cycloheptane_2', 'cyclooctane_1',
'cyclooctane_2', 'diaminobenzene_1', 'diaminobenzene_2',
'diaminobenzene_3', 'diazo_group', 'diketo_group', 'disulphide',
'enamine', 'ester_of_HOBT', 'four_member_lactones',
'halogenated_ring_1', 'halogenated_ring_2', 'heavy_metal',
'het-C-het_not_in_ring', 'hydantoin', 'hydrazine', 'hydroquinone',
'hydroxamic_acid', 'imine_1', 'imine_2', 'iodine', 'isocyanate',
'isolated_alkene', 'ketene', 'methylidene-1,3-dithiole',
'Michael_acceptor_1', 'Michael_acceptor_2', 'Michael_acceptor_3',
'Michael_acceptor_4', 'Michael_acceptor_5', 'N_oxide',
'N-acyl-2-amino-5-mercapto-1,3,4-_thiadiazole', 'N-C-halo',
'N-halo', 'N-hydroxyl_pyridine', 'nitro_group', 'N-nitroso',
'oxime_1', 'oxime_2', 'Oxygen-nitrogen_single_bond',
'Perfluorinated_chain', 'peroxide', 'phenol_ester',
'phenyl_carbonate', 'phosphor', 'phthalimide',
'Polycyclic_aromatic_hydrocarbon_1',
'Polycyclic_aromatic_hydrocarbon_2',
'Polycyclic_aromatic_hydrocarbon_3', 'polyene',
'quaternary_nitrogen_1', 'quaternary_nitrogen_2',
'quaternary_nitrogen_3', 'saponine_derivative', 'silicon_halogen',
'stilbene', 'sulfinic_acid', 'Sulfonic_acid_1', 'Sulfonic_acid_2',
'sulfonyl_cyanide', 'sulfur_oxygen_single_bond', 'sulphate',
'sulphur_nitrogen_single_bond', 'Thiobenzothiazole_1',
'thiobenzothiazole_2', 'Thiocarbonyl_group', 'thioester',
'thiol_1', 'thiol_2', 'Three-membered_heterocycle', 'triflate',
'triphenyl_methyl-silyl', 'triple_bond'], dtype='<U44')
Underlying SMARTS patterns are represented using RDKit FilterCatalog objects, which are quite efficient in checking the patterns. Unfortunately, getting the actual SMARTS strings in Python is challenging, and more easily inferred from RDKit files. The first few patterns from BRENK and their meanings are:
[85]:
patterns = [
(">_2_ester_groups", "C(=O)O[C,H1].C(=O)O[C,H1].C(=O)O[C,H1]"),
("2-halo_pyridine", "n1c([F,Cl,Br,I])cccc1"),
("acid_halide", "C(=O)[Cl,Br,I,F]"),
("acyclic_C=C-O", "C=[C!r]O"),
("acyl_cyanide", "N#CC(=O)"),
("acyl_hydrazine", "C(=O)N[NH2]"),
("aldehyde", "[CH1](=O)"),
("Aliphatic_long_chain", "[R0;D2][R0;D2][R0;D2][R0;D2]"),
]
[86]:
from rdkit.Chem import Draw
mols = []
titles = []
for name, smarts in patterns:
mol = Chem.MolFromSmarts(smarts)
if mol is not None:
mols.append(mol)
titles.append(name)
img = Draw.MolsToGridImage(mols, legends=titles, molsPerRow=4, subImgSize=(300, 300))
img
[86]:
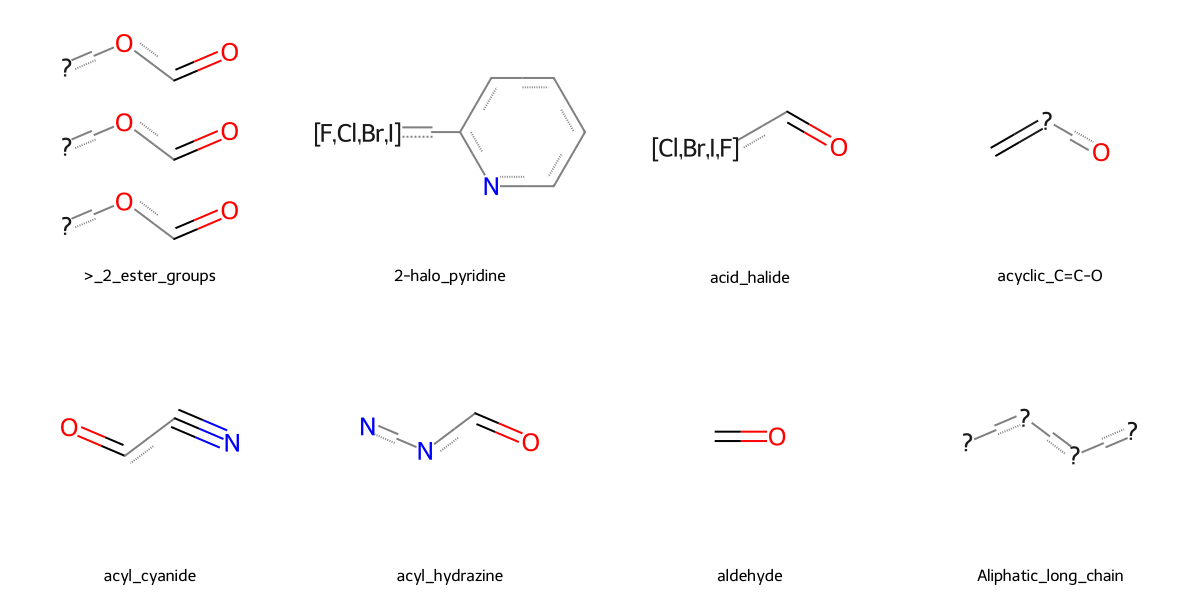
Let’s focus on aldehydes. One of the simplest aldehydes is acrolein. Let’s see its structural formula.
[100]:
acrolein_smiles = "C=CC=O"
mol = Chem.MolFromSmiles(acrolein_smiles)
img = Draw.MolToImage(mol, size=(300, 300))
img
[100]:
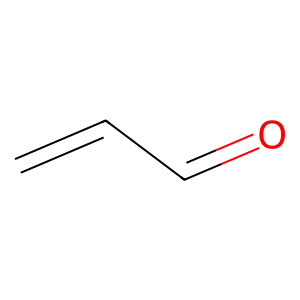
Obviously, it has a carbonyl group, which is characteristic of aldehydes. Thus, it should be filtered out by the Brenk filter.
[101]:
brenk_filter = BrenkFilter()
smiles = [acrolein_smiles]
filtered_smiles = brenk_filter.fit_transform(smiles)
filtered_smiles
[101]:
[]
Custom filters#
By inheriting from BaseFilter, you can create your own molecular filters. The main element is the _apply_mol_filter method, which should return True if a molecule fulfills the filter rules and should be kept, or False otherwise.
Let’s create a simple physicochemical filter, which will keep molecules below max_weight and having at most num_heavy_atoms heavy atoms. This is a slightly simplified example, without fully-featured parameter validation - see scikit-learn code if you want to learn more about that.
Further, all filters have arguments:
allow_one_violation- if you want to tolerate a single violationreturn_indicators- whether to return molecules or a boolean maskn_jobs- how many processes to use for filtering in parallelbatch_size- for finer control over batch size for parallelismverbose- controls verbosity of TQDM progress bar
You can omit those arguments, but it will also disable some of the functionalities that you can expect from scikit-fingerprints filters.
[100]:
from rdkit.Chem import Mol
from rdkit.Chem.Descriptors import MolWt
from rdkit.Chem.rdMolDescriptors import CalcNumHeavyAtoms
from skfp.bases.base_filter import BaseFilter
class CustomMolecularFilter(BaseFilter):
def __init__(
self,
max_weight: int = 250,
num_heavy_atoms: int = 50,
allow_one_violation: bool = False,
return_indicators: bool = False,
n_jobs: int | None = None,
batch_size: int | None = None,
verbose: int | dict = 0,
):
super().__init__(
allow_one_violation=allow_one_violation,
return_indicators=return_indicators,
n_jobs=n_jobs,
batch_size=batch_size,
verbose=verbose,
)
# create attributes from custom parameters
self.max_weight = max_weight
self.num_heavy_atoms = num_heavy_atoms
def _apply_mol_filter(self, mol: Mol) -> bool:
# define the filter rules and check them
rules = [
MolWt(mol) <= self.max_weight,
CalcNumHeavyAtoms(mol) <= self.num_heavy_atoms,
]
# calculate how many rules have passed
passed_rules = sum(rules)
# check final filter status
if self.allow_one_violation:
return passed_rules >= len(rules) - 1
else:
return passed_rules == len(rules)
[101]:
smiles = [
"CC(=O)OC1=CC=CC=C1C(=O)O", # Aspirin
"CN1C=NC2=C1C(=O)N(C(=O)N2C)C", # Caffeine
"CC(C)CC1=CC=C(C=C1)C(C)C(=O)", # Ibuprofen
"C[C@H](CCC(=O)O)CC1=CC=CC=C1", # Cholesterol
]
[102]:
custom_filter = CustomMolecularFilter(verbose=True)
[103]:
custom_filtered_smiles = custom_filter.fit_transform(smiles)
custom_filtered_smiles
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 4/4 [00:00<00:00, 22919.69it/s]
[103]:
['CC(=O)OC1=CC=CC=C1C(=O)O',
'CN1C=NC2=C1C(=O)N(C(=O)N2C)C',
'CC(C)CC1=CC=C(C=C1)C(C)C(=O)',
'C[C@H](CCC(=O)O)CC1=CC=CC=C1']
[ ]: